Malformazione Adenomatoide Cistica Congenita
Chirurgia Pediatrica
Cosa è?
Con il termine di Malformazione Adenomatoide Cistica Congenita (o malformazione congenita delle vie aeree polmonari o l’acronimo CCAM ovvero Congenital Cystic Adenomatoid Malformation), si intendono un insieme di malformazioni polmonari, caratterizzati dalla presenza di una o più cisti polmonari, di dimensioni variabili, circondate da tessuto polmonare sano.
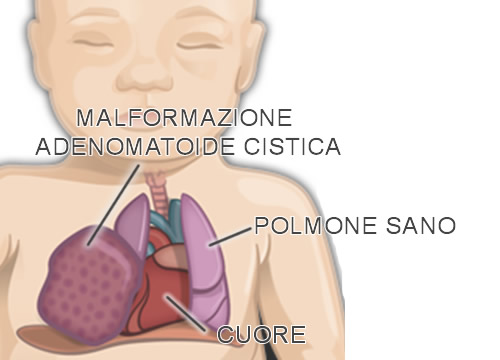
È una patologia congenita data da un anomalo sviluppo degli alveoli, ovvero l’unità funzionale dei polmoni, in cui l’aria arriva, per mezzo delle vie aeree, e l’ossigeno, contenuto in essa, viene trasferito al sangue. In questa malformazione una parte degli alveoli non si sono sviluppati e sono stati sostituiti da delle strutture cistiche, in continuità con le vie aeree e la cui parete è costituita da una varietà di tessuti, che non risultano funzionali all’ossigenazione del sangue.
Questa patologia solitamente interessa solo uno dei polmoni e alle volte può trovarsi associata ad altre anomalie.
Vi sono diversi modi di classificare la CCAM, fra cui uno dei più semplici, si basa sulle dimensioni e sulla diffusione delle cisti, identificandone tre tipi:
- Tipo 1, caratterizzato dalla presenza di una o più cisti le cui dimensioni superano i 2 cm. Questa rappresenta la tipologia più comune;
- Tipo 2, caratterizzato dalla presenza di cisti multiple molto piccole, ovvero al di sotto dei 2 cm. Questa rappresenta la tipologia solitamente associata ad altre anomalie.
- Tipo 3, caratterizzata dalla presenza di una lesione piuttosto grande di dimensioni, occupante un lobo polmonare, e a contenuto solido e non liquido;
Quali sono i sintomi?
Raramente è possibile identificale la sintomatologia, in quanto i pazienti affetti da CCAM, sono identificati in epoca prenatale, rendendo possibile un parto e delle cure perinatali congrui.
La sintomatologia è in relazione con il tipo di malformazione, infatti il secondo e il terzo tipo sono spesso associati a importanti difficoltà respiratorie alla nascita, mentre il primo tipo può rimanere asintomatico per un cero periodo di tempo e divenire manifesto tardivamente, per il sopraggiungere di infezioni respiratorie ricorrenti o per la rottura di una delle cisti che comporta la liberazione di area nella gabbia toracica (pneumotorace), rendendo difficoltosa la respirazione.
Come si giunge alla diagnosi?
La diagnosi è posta generalmente in epoca prenatale, per mezzo dell’ecografia, che ci consente di identificare direttamente la lesione o i segni indiretti della sua presenza, fra cui un polmone omolaterale di ridotte dimensioni, uno spostamento controlaterale del cuore e un aumento del volume del liquido amniotico. La diagnosi prenatale è di fondamentale importanza per valutare l’evoluzione della patologia in utero e scegliere il timing del parto, qualora la cisti diventasse troppo grande, e per indirizzare i genitori in un centro in cui sia presente un’unità operativa di terapia intensiva neonatale e una di chirurgia pediatrica, che possano prendere in carico il piccolo fin dalla nascita, garantendo supporto ventilatorio e procedure chirurgiche immediate, qualora richieste.
In epoca post natale la diagnosi può essere posta invece con l’Rx del torace che permette di individuare o delle anomalie generiche del tessuto polmonare, o le lesioni cistiche direttamente o uno spostamento laterale del cuore.
Tuttavia, qualora vi sia un sospetto di CCAM è sempre bene eseguire una TC del torace, intorno ai 4/6 mesi, in quanto questa ci consente di studiare in maniera accurata l’anatomia polmonare e di valutare la presenza della malformazione.
Quando e perchè è necessario il trattamento chirurgico
Il trattamento chirurgico è l’unica opzione terapeutica che ci consente di evitare lo sviluppo di difficoltà respiratorie e di complicanze a lungo termine, prime fra tutti lo sviluppo di infezioni respiratorie ricorrenti e la trasformazione in neoplasia, infatti esiste un consensus internazionale, che evidenzia la trasformazione neolpastica della CCAM, qualora questa non fosse trattata in maniera adeguata.
Il timing del trattamento chirurgico dipende dalle condizioni cliniche del piccolo, infatti nel caso di importanti difficoltà respiratorie, il piccolo andrà incontro all’intervento nella maniera più tempestiva possibile, nel caso di condizioni cliniche favorevoli, il trattamento chirurgico potrà, invece, essere rimandato ai 18 mesi di vita del piccolo.
In cosa consiste
Il trattamento chirurgico consiste nella resezione della lesione e del lobo polmonare interessato. Tale procedura può essere eseguita o con approccio mininvasivo, ovvero attraverso delle piccole incisioni utili ad introdurre una telecamera e gli strumenti, o con approccio tradizionale, ovvero attraverso un’incisione più ampia in sede postero-laterale del torace. Raramente si dovrà ricorrere a resezioni più ampie di tessuto polmonare.
Inoltre qualora la prima manifestazione clinica sia lo pneumotorace, sarà necessario far fuoriuscire l’aria dalla gabbia toracica, in maniera tempestiva, per mezzo di un drenaggio toracico, che servirà solo a migliorare la capacità respiratoria e consentire di giungere al trattamento definitivo, nelle migliori condizioni possibili.
Quanto tempo rimane in ospedale il piccolo?
La permanenza del piccolo in ospedale, dipende da tanti fattori, primi fra tutti il riscontro della malformazione in epoca prenatale, le caratteristiche della malformazione e le condizioni cliniche del piccolo. In generale un bambino con diagnosi prenatale e riscontro di condizioni cliniche non soddisfacenti, rimane in ospedale dalla nascita, fino al trattamento chirurgico, dopo il quale la degenza è di qualche settimana, in quanto sarà necessario eseguire uno stretto monitoraggio delle condizioni cliniche del piccolo, la somministrazione in vena di antibiotici e l’esecuzione di medicazioni apposite.
Quale sarà la qualità di vita del piccolo?
La qualità di vita del piccolo sarà uguale a quella della popolazione sana, qualora sia stato possibile asportare in toto la lesione e assicurare la permanenza di tessuto polmonare sufficiente alla necessità del paziente.

Navigazione